
What Is Human Factors and When to Start?

When to incorporate human factors into your regulatory strategy can have a serious impact on the product development timeline and budget, planned submission date, and submission success rate.
All too often clients approach human factors late in the product development process, this approach can result in a negative impact on budget due to delayed submission to the regulatory agency. Human factors testing is a premarket requirement and is most effective when started early in product development. So why do sponsors wait so long to engage in human factors testing? I believe there are several reasons including a lack of understanding of what human factors is and best practices in application.
What is Human Factors?
Human factors engineering focuses on the interactions between people and user interfaces. Human factors studies are qualitative in nature and evaluate the safety and effectiveness of the medical product when in the hands of the user.
For medical devices, the most important goal of the human factors engineering process is to optimize the user interface through minimizing use-related hazards and risks and then confirming that these efforts were successful by verifying users can use the device safely and effectively. The user interface includes all device components the user interacts with to prepare the device for use, use the device, or perform maintenance on the device.
There are various human factors research methods including simulated use or human factors studies. Additionally, there are different types of human factors studies:
Early formative studies typically use a smaller sample size of 5-8 users per group, may evaluate focused aspects of the user interface rather than the entire user interface, and may evaluate design alternatives and early concepts, or user characteristics and preferences, environmental characteristics to inform design assumptions, device design or selection.
Late-stage formative studies bring more aspects of the user interface together to evaluate at one time, eventually testing the entire user interface and approaching commercial design and validation study methodology. Late-stage formatives also inform design of device, packaging, instructions and or training, and build readiness for a successful validation.
Summative or validation studies use a larger sample size of at least 15 users per group, assess the entire user interface with all defined user groups, use the final commercial-ready design of the device or product user interface, and demonstrates use related safety and effectiveness of the device or product user interface in the hands of the end users. Human factors studies are not marketing research, preference studies or clinical studies. Ideally, HF validation testing should precede any major clinical study to support the use of the proposed product by the intended end users in the clinical study. In some cases, it may be appropriate to conduct HF studies in parallel or after clinical studies.
Why we need Human Factors?
Human factors in regulatory testing is a growing industry and required by regulatory health authorities around the world.
US regulatory requirements for human factors are embedded within the Code of Federal Regulations, Quality System Regulations Design Controls (US 21CFR 820.30). Regulators are aware of thousands of reports each year detailing the deaths, serious injuries and malfunctions associated with medical devices. This creates increased regulator attention to human factors on new submissions. According to a recent FDA study[1], 90% of first time submissions are labeled incomplete.
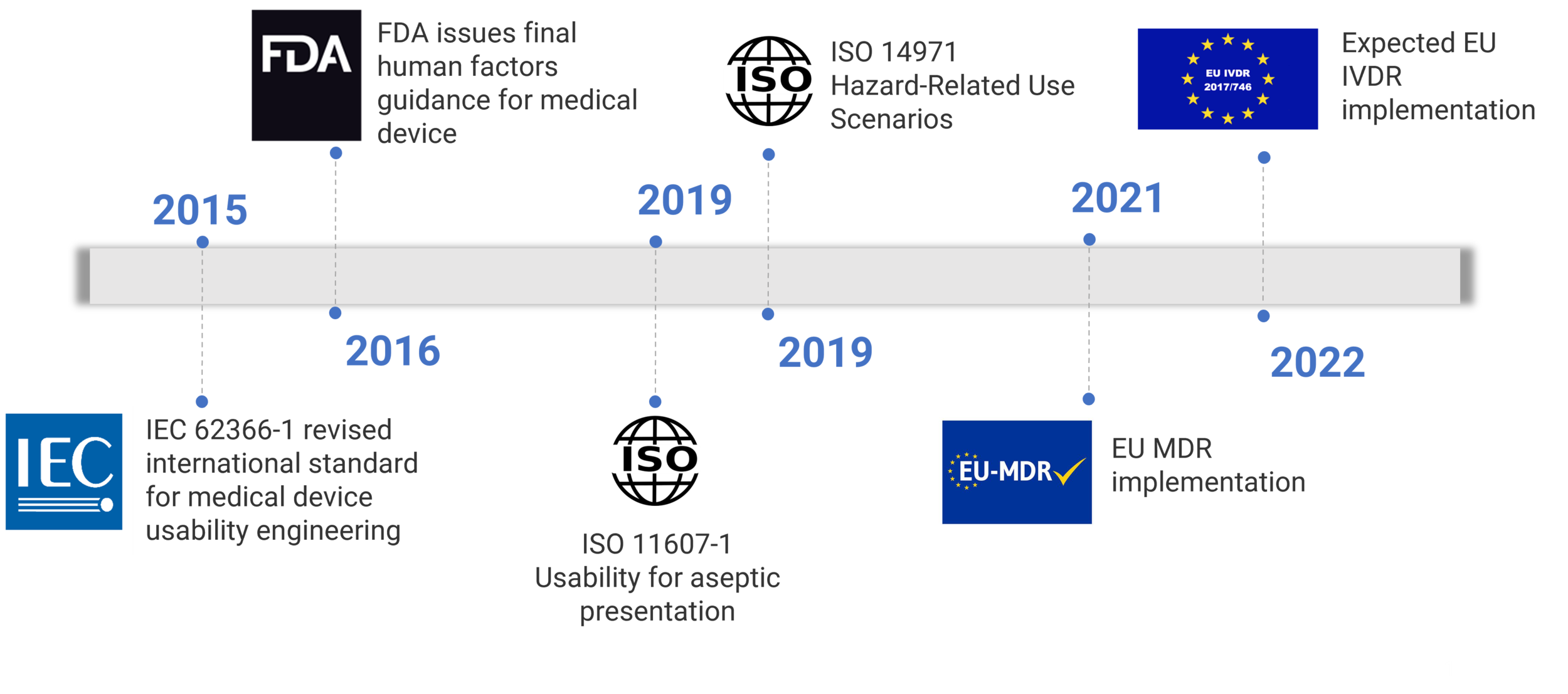
Figure 1: Regulatory growth timeline
When to start Human Factors?
With the goal of the human factors engineering process being to minimize use-related hazards and risks and to ensure users can use the device safely and effectively, the ideal time to include human factors engineering is early enough in the product development process to impact product design and the user interface to reduce use-related risks.
Delays occurring just before or within a submission are costly and may require protocol revision and repeated study conduct or a supplemental validation study. The result of which can add six months or more to your timeline and cost hundreds of thousands of dollars, upending your budget.
In this case study, for a combination product device, it will demonstrate what can happen when a human factors strategy is not integrated early in the product development process.
Company Strategy: Sponsor company was developing a combination product inhaler. The sponsor company engaged a device partner without human factors assessment of the device with intended users in the intended environment. The sponsor company sought regulatory interaction early but changed methodology late in the submission timeline. Late-stage interactions included:
Pre-submission meetings with FDA CDER/DMEPA
Pre-submission review of human factors validation protocol
Pre-submission review of commercial labeling and packaging

Figure 2: Pay Later approach
Company Result: The initial validation study included all trained participants. During the FDA review of HFE/UE report, CDER asked for human factors data for untrained participants because, “You indicated that the training will be ‘offered’ to the patient but there is no assurance that all patients are trained.” This feedback of subsequent FDA reviews was received from the Agency after formative studies were conducted.
The client then called Agilis to re-design the IFU, iterate preliminary analyses and conduct validation study because changes to the device design were not an option.
Selecting your partner: Implications
FDA did a natural language processing search and analysis on the submissions reviewed during the course of 2019[1]. The results of this analysis show that, based on submission type, the human factors submissions accepted the first time range from only 4-11%.

Figure 3: URRA Impacts on CDRH clearance or approval
This data means that the implications of choosing the right human factors partner can greatly impact your medical device/product timeline and budget. No matter the human factors partner you work with there are certain strategies that could result in a pay later model:
Limited or no engagement with regulatory authority, causing no feedback on risk assessment or human factors strategy.
No human factors testing included in the product development timeline and the regulatory timeline or only a validation/summative study is planned.
No early human factors formative evaluations to optimize design of the user interface prior to validation/summative study. Hard submission date with no flexibility to optimize design.
No human factors data to support responses to regulatory authority during the submission review process.
Each human factors study could reveal that changes are necessary to the user interface, this could be the device, packaging, instructional materials, or training. The project timeline or submission schedule should not only account for the studies, but also include opportunities to revise the schedule to accommodate design changes.
If human factors is not included early, there can be a compression to the timeline that makes it more difficult to make decisions about design changes and the planned submission date.
If manufacturing is already being established (later in product development), design changes become very difficult to make. Prototyping stage is better for making design changes and optimizing the design of all aspects of the user interface.
With this data, many of our clients have established an internal case for pushing the submission date. Not all clients do this and sticking to a hard date can still mean post-submission delays.
Making design changes after manufacturing processes are established and validated.
Delivery device not being suited to your product or patients – where do you go from here?
Increases in timeline and budget and extending submission date beyond what was planned.
Delays in timeline to obtain regulatory authority input.
Little to no human factors data to build a human factors story about the optimization of your product/device and reduce the risk of requests for additional information after submission.
Conclusion
The best strategy is to account for human factors execution in the product development timeline and the regulatory timeline. This includes engaging with the regulatory authority through pre-submission meetings to confirm a regulatory pathway and related human factors guidance, as well as to receive feedback on the risk assessment and human factors protocol for agreement on user groups and testing methods. Results of human factors studies could impact the regulatory strategy and submission timeline. Therefore, it is best to integrate human factors strategies early in the product development process.
Agilis partners with our clients to provide end users with safer and more reliable medical products and we are proud of our success rate!
The FDA has never rejected the Human Factors methodology from an Agilis client!
References:
[1]Wiyor, H.D. (October 22, 2020). Risk Assessment of Medical Products in Human Factors Submissions with a Focus on EU countries. MHFN Webinar.
Additional Sources
US Department of Health and Human Services Food and Drug Administration. (2016, February 3). Applying Human Factors and Usability Engineering to Medical Devices. Guidance to Industry and Food and Drug Administration Staff. Retrieved December 7, 2017, from: https://www.fda.gov/downloads/MedicalDevices/.../UCM259760.pdf
US Department of Health and Human Services Food and Drug Administration. (2016, February 3). List of Highest Priority Devices for Human Factors Review. Draft Guidance for Industry and Food and Drug Administration Staff. Retrieved December 7, 2017, from: https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-meddev-gen/documents/document/ucm484097.pdf
US Department of Health and Human Services Food and Drug Administration. (2016, February). Human Factors Studies and Related Clinical Study Considerations in Combination Product Design and Development. Draft Guidance for Industry and FDA Staff. Retrieved December 7, 2017, from: https://www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM484345.pdf
About the Author:
Agilis Team

Agilis’ team are all seasoned human factors experts, many of whom are considered industry thought leaders. We strive to maintain consistency of your Agilis team throughout the project lifecycle. Our unabashed commitment to quality is evidenced by a rigorous internal quality control system including on-going professional development for all Agilis team members.