
Back to Basics – Human Factors Engineering Process for Medical Device Products

Clients and Colleagues in the healthcare, medical device, pharmaceutical and life science industries:
Happy 2022! I decided to start this new year by revisiting the basics. What I’d like to do is revisit the Human Factors/Usability Engineering process for Medical Device products. (Yes, even those that are part of a combination product.) For this activity, I am pulling out one of my favorite books: The Quality Toolbox, 2nd edition; authored by Nancy R. Tague and published by ASQ. From this toolbox, the goal was to map the HFE/UE process using a SIPOC (suppliers-inputs-process-outputs-customers) diagram. Using the international standard AAMI/ANSI/IEC 62366-1:2015+AMD1:2020 framework, I built a model for discussion.
DISCLAIMERS:
Every design control process uses different document names and terminologies; however, the core building blocks should be consistent, if not by name then by intent. An official Design Review by any other name would still have an independent reviewer, as such.
Admittedly this is not all inclusive and is more of a brushstroke view of product development. I would love to hear from you what other items should be accounted for, so please respond below.
TLA – Acronyms, we all love them. Here are some that I use:
CEUD – Context of Expected Use Document
DDP – Design & Development Plan
GSPR – General Safety and Performance Requirements (MDR/IVDR)
HA – Hazard Analysis
HFE/UE Report - as a specific FDA submission document
R&D – Research & Development (core development team)
RA/QA – Regulatory Affairs/ Quality Assurance
RMF – Risk Management File
URRA – Use-related Risk Assessment
V&V – Design Verification & Validation
With all that said, here is a SIPOC for the Human Factors process. I am going to share this graphic twice. First, as a mapping of the IEC 62366-1 framework, as shown in Figure 1.
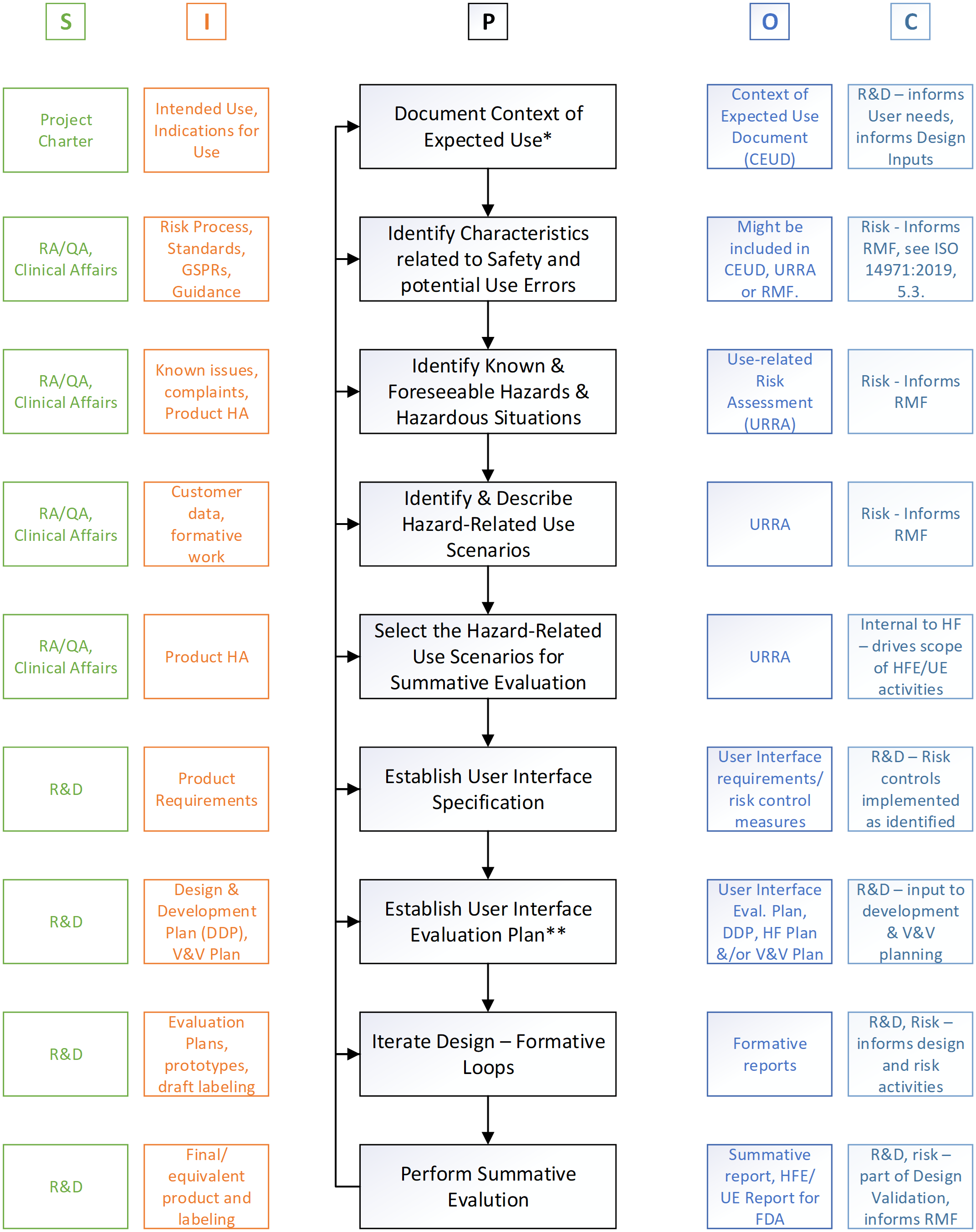
Figure 1: SIPOC for the Human Factors process © Agilis Consulting Group
I would like to comment on a few points before we proceed. First (*) what I call the Context of Expected Use. This, in the standard, is referred to as the Use Specification. I have found, at least in English, this term ‘specification’ causes quite a bit of consternation. So, my suggestion is to keep the spirit of it, and build the document to capture the information that you need in order to define the context around the use (use scenarios, users, use environments, etc.) of your product that is necessary to inform and focus the development activities. In my opinion this is an informational document not a requirements document (or specification document).
Second point (**) for discussion, the user interface evaluation plan. Rather than a stand-alone document, I have seen this be part of and an update to the HF plan, if you have one; or, it could be part of a larger product evaluation plan. Regardless of where it lives the intent outlined in the standard should be captured.
Figure 1 outlines the IEC 62366-1 process in a SIPOC diagram; but I would also like to take a look at it through the lens of the FDA Human Factors Guidance (2016 Final) and MDR/GSPR perspective. Some of these considerations are captured in Figure 2.


Figure 2: SIPOC for the Human Factors process part 2 © Agilis Consulting Group
Please, if you are still reading this, allow me to elaborate on my thought process. Starting with the FDA 2016 Final HF guidance.
Conclusion – this sums up that the body of evidence in the report substantiates a conclusion that the User Interface supports safe and effective use for the intended users, uses and use environments. This is going to rely on the body of data from the HF process, identified use-related risks and their relationship to the benefit-risk for your product.
Description of intended device users, uses, use environments and training – this information can be lifted from the Context of Expected Use Document, which is maintained throughout the iterative development and HF activities.
Description of the device user interface – this may live in the HF documentation, or it may live in a product or system level document.
Summary of known use problems, analysis of hazards and risks, and categorization of critical tasks – will likely be attributed to the URRA activities which evolve over the development (and product) life cycle.
Summary of preliminary analyses and evaluations – will summarize formative activities conducted to inform the design and URRA.
Human Factors Validation – all of the information in sections 2-7, define the scope of the Human Factors Validation activities. Section 8 should provide details on the scope and representative-ness of the study design, using sound methodology to collect objective and subjective data and provide the analysis of said data.
A few points of interest with regards to the GSPR’s:
For Chapter 1, consider how the HFE/UE process could provide a methodology and evidence to support the requirements outlined in items 5a and 5b.
For Chapter 2, within this chapter there are many items outlined that could be hazards or could contribute to hazards/hazardous situations. I suggest that this whole list be considered when identifying the characteristics related to safety. Doing this early in the process will also help you to capture, track and evaluate specific requirements such as:
11.4 Devices delivered in a sterile state shall be designed, manufactured and packaged in accordance with appropriate procedures, to ensure that they are sterile when placed on the market and that, unless the packaging which is intended to maintain their sterile condition is damaged, they remain sterile, under the transport and storage conditions specified by the manufacturer, until that packaging is opened at the point of use. It shall be ensured that the integrity of that packaging is clearly evident to the final user.
For Chapter 3, this contains requirements for the information supplied with the device and is supported by the HF process when the instructions and information are considered part of the user interface developed within the HF process. Note, that the inclusion to this information, packaging and training provided with the product are also in scope per the standard and the 2016 FDA guidance. As such these requirements should also inform the various process activities including the evaluation plan and summative activities.
Whew! This overview was a good reminder of how our HF work integrates into product development and regulatory submission. I hope that it is helpful for you whether you are newer to human factors or mired in it.
At Agilis, we very much appreciate having had the opportunity to serve our clients’ HF needs in 2021 and look forward to helping you, wherever 2022 finds you in this process.
About the Author:
Shannon Hoste

Shannon leads the high-performance team of experienced human factors, biomedical engineers and instructional design experts to support Agilis’ US and global clients from early-stage user research, through regulatory submission and post-market surveillance. She is an expert in human factors and product development processes with over 20 years of experience in the industry.
AAMI Book

Applied Human Factors in Medical Device Design